


After more than two decades, the legal architecture governing medicinal products in the European Union is being fundamentally rebuilt. Directive 2001/83/EC and Regulation (EC) No 726/2004 are being replaced by a new Regulation and a new Directive that together redefine the terms on which medicines are authorised, protected, and made available across the Union.
The legislative process is nearly finished, as on 6 March 2026[1], the European Council published its "compromise texts of the provisional agreement" (the "Compromise Texts" or the "new Regulation" and the "new Directive").[2] Given the advanced stage of the legislative process, substantive changes to the legislative texts are now considered unlikely.
Accordingly, the focus should shift from monitoring the legislative process to preparing for and operationalising the new framework. This article maps that transitional architecture of the reform in detail, with a focus on the concrete operational decisions it forces on pharmaceutical companies.
1. Adoption, Entry into Force, Application
The political agreement between the European Council and the European Parliament was reached in December 2025, following close to two years of trilogue negotiations since the Commission's original proposal of April 2023.[3] The Compromise Texts are dated 24 February 2026 and were published in March 2026. These texts represent the agreed-upon legislative substance, but they are not yet formally adopted law.
To become applicable binding law, the texts need to be formally adopted by the European Council, which will happen only after they have passed a formal vote in the European Parliament. On the current trajectory, formal adoption is expected in Q4 of 2026, though no definitive date has been confirmed at the time of writing.
After its adoption, the legislation will be published in the Official Journal of the European Union ("OJEU"). The new Regulation and new Directive will enter into force on the twentieth day following that of their publication in the OJEU. The entry into force is the date on which the act becomes legally effective as a matter of EU law.
However, the Compromise Texts include transitional provisions that postpone the concrete application of the most substantive parts of the texts to a later point in time, allowing all stakeholders to prepare for and adapt to the new legislation. The texts will only produce legal consequences after the date of application.
We will first present the key takeaways of the transitional regime and will analyse the provisions in depth afterward:
2. Key Takeaways of the Transitional Regime for Pharmaceutical Companies
Some important provisions will apply immediately[4] or shortly after[5] the publication of the acts and should therefore already be taken into consideration now:
- Direct application of the Transferable Exclusivity Voucher ("TEV"): The provisions on the TEV incentive already apply as of the entry into force of the revised pharmaceutical legislation, which is expected for Q4 2026. This means that companies developing antimicrobials must start the process to compile a dossier to be able to apply immediately. It also means that the exclusivity vouchers could be used to extend existing marketing protection periods gained under the current regime.
- Supply obligations: The security of supply and critical medicines framework is immediately operative upon entry into force. The shortage notification and the obligatory shortage prevention plans follow six months later. The obligation to have a shortage prevention plan in place will also explicitly apply to products authorised under the old Regulation and old Directive before the new Regulation's application date. The plans should therefore be ready and operational by mid-2027 for the whole product portfolio.
- Supply conditionality mechanism: From twelve months after entry into force of the new Directive, Member States ("MS") may invoke the launch and supply conditionality mechanism for products authorised after that date. As the rest of the provisions only apply after 24 months, the conditionality mechanisms can lead to situations where companies lose market protection that was granted under the old regime.
The vast majority of the provisions of the Directive and the new Regulation will only apply 24 months after their entry into force. Their application will have the following impact on pending procedures and portfolios:
- The submission date determines the regulatory protection regime: Medicinal products whose reference Marketing Authorisation ("MA") application is filed before the application date of the new Regulation or new Directive retain the old data exclusivity and market protection rules. Medicinal products filed on or after that date fall under the new exclusivity regime.
- Orphan market exclusivity grandfathering is limited to the initial MA: Orphan MAs applied for before the date of application benefit from continued old-regime exclusivity. However, this is only true for the initial or first MA granted for a certain active substance. Subsequent applications for the same active substance and with a separate orphan designation, even if filed before the application date, fall under the new regime, which means that they will not give rise to a new full period of market exclusivity, but will follow the duration of market exclusivity of the first MA granted for that active substance.[6]
- Pending procedures are generally complete under the old rules, with one exception: pending centralised MA applications, national/DCP/MRP applications, PIPs, and referrals initiated before the application date are completed under the old regime. The exception is pending post-authorisation studies (PASS), which switch to Article 20 of the new Regulation on its date of application.
- Renewals falling due after the application date follow the new procedural rules: Companies should audit renewal calendars for the 24 to 48 months around the expected application date and ensure dossier preparation reflects the requirements of Article 17 of the new Regulation and Article 46 of the new Directive respectively.
3. The New Regulation
In principle, the new Regulation shall enter into force on the twentieth day following that of its publication in the Official Journal of the European Union. However, Article 181 of the new Regulation provides that some provisions will apply only 6 months after the entry into force, and that the vast majority of the provisions will only apply from 24 months after the entry into force. The new Regulation will therefore apply in three phases:[7]
- Phase 1: Publication in the OJEU + 20 days.
- Phase 2: Publication in the OJEU + 20 days + 6 months.
- Phase 3: Publication in the OJEU + 20 days + 24 months.
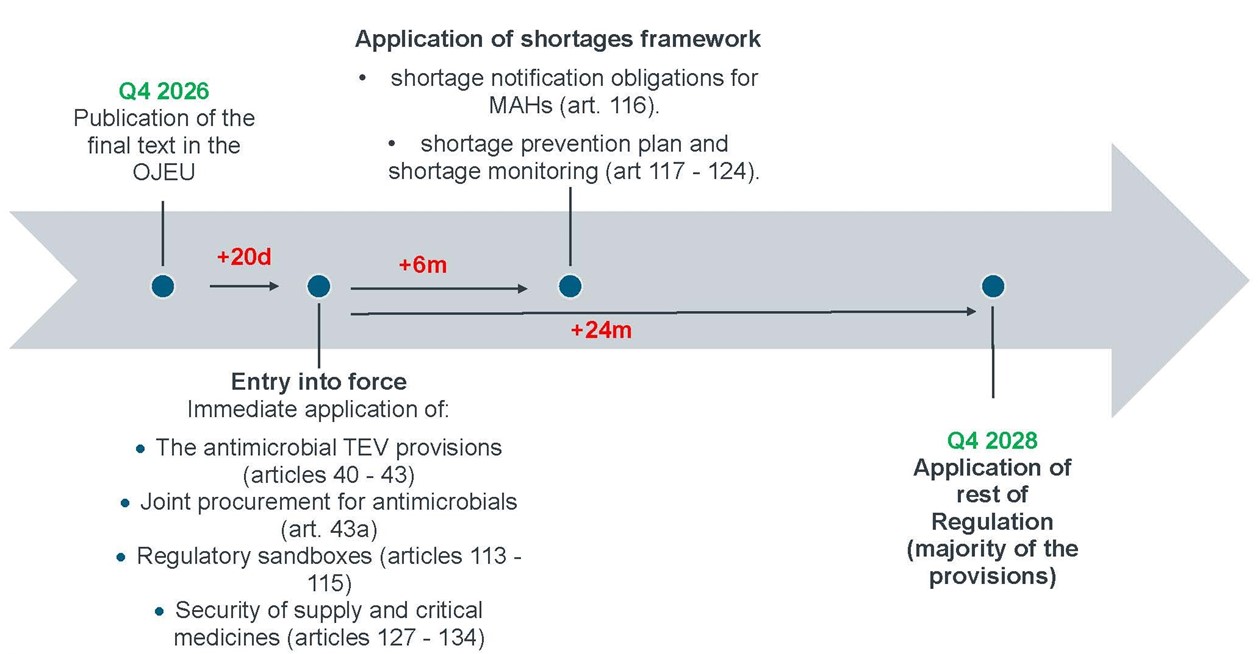
3.1 Phase 1: Publication in the OJEU + 20 Days
In the first phase, and thus 20 days after the publication in the OJEU of the Regulation, the following provisions immediately apply:[8]
- The antimicrobial TEV provisions (Articles 40–43): The TEV gives its holder the right to an additional 12 months of regulatory data protection for one priority antimicrobial.[9] Companies that have or are developing qualifying antimicrobials can therefore begin engaging with the criteria for designation and voucher grant before the bulk of the new regulatory exclusivity regime applies. This includes checking whether the mechanism of action of their antimicrobials is distinctly different from that of any authorised antimicrobial in the EU or if they contain a new active substance that, when used either alone or in combination with other active substances, addresses a serious or life-threatening infection. It also means that the exclusivity vouchers could be used to extend existing marketing protection periods gained under the current regime. Companies that will have an early TEV will therefore have a valuable asset on the market.
- Joint procurement for antimicrobials: The joint procurement model of Article 43a that allows contracting authorities from different Member States ("MS") to act jointly in the award of public contracts for the purchase of antimicrobials will be live already from the beginning. This novelty is not groundbreaking as the option to organise cross-border joint procurements is already provided by the general directive on public procurement.[10] What is new is that the different MS may request the Commission to organise joint meetings between the competent authorities of the MS, and any other relevant parties, as appropriate, including bodies responsible for pricing and reimbursement, to discuss issues related to the practical application of the joint procurement.
- Regulatory sandboxes (Articles 113–115): The framework enabling structured regulatory experimentation for innovative products applies immediately, allowing early engagement with competent authorities under the new mechanism.
- Security of supply and critical medicines (Articles 127–134): The Union-level architecture for identifying critical medicinal products, conducting vulnerability evaluations, and triggering coordinated responses is operative from entry into force. This includes the Union list of critical medicinal products, obligations of marketing authorisation holders ("MAHs") regarding critical medicinal products, and the Commission's and the European Medicines Agency's ("EMA") powers to request information from MAHs on products that feature on those lists.
3.2 Phase 2: Publication in the OJEU + 20 Days + 6 Months
In the second layer, the operational core of the shortage monitoring and MAH obligations relating to shortage management will become applicable. With the security of supply and critical medicines framework already in application, this gives the following situation after 6 months:
- The shortage notification obligations for MAHs (Article 116). Under this Article, MAHs will have to notify the EMA and explain the reasons for their decision to withdraw or suspend the marketing of a product. This obligation will therefore be applicable to products authorised under the old regime.
- The provisions that introduce a mandatory shortage prevention plan and shortage monitoring by the competent authorities (Articles 117–118). The preparation for drafting a shortage prevention plan should therefore already start now. The shortage prevention plan should include shortage prevention measures and supply chain risk assessment, such as an estimation of the patient impact of potential supply disruptions, considering therapeutic indication and alternative marketed medicinal products and estimated market share by Member States in the previous 12 months.[11]
Article 117 of the new Regulation, which introduces the obligation to have a shortage prevention plan in place, will apply to MAs granted under the old regime of Regulation (EC) No 726/2004 and Directive 2001/83/EC before the Regulation's application date. These plans should thus be ready by mid-2027 for the entire product portfolio, not only for medicinal products that received an MA under the new regime.
- Monitoring obligations for MAHs and distributors, MAH cooperation duties with the competent authorities, the structured information exchange between national competent authorities and EMA and the role of the MSSG[12] (Articles 119–124).
- Information obligations on the MAH in case of a critical shortage of Union concern (Article 125), the voluntary solidarity mechanism for medicinal products which allows Member States to request surplus medicines from other countries (Article 125a) and the role of the Commission in the process (Article 126).
3.3 Phase 3: Publication in the OJEU + 20 Days + 24 Months
The rest (and thus the vast majority) of the Regulation will apply from 24 months after its entry into force. From this date, the full procedural architecture will become applicable, such as the new centralised MA process, the new variation and renewal regime, the new regulatory protection framework, and the new orphan and paediatric incentive. Regulations (EC) No 141/2000, (EC) No 726/2004 and (EC) No 1901/2006 and implementing Regulation 198/2013 will be repealed on this date.[13]
Apart from the three phases above, we must mention that Article 67, which introduces the adoption of a register of designated orphan medicinal products, will only apply 30 months after the entry into force.
3.4 Impact of Application on Pending Applications and Procedures
The transitional provisions determine whether a pending procedure under the old regulatory regime or an application made before the entry into force of the new Regulation is governed by the new Regulation or by the old regime.
3.4.1 Pending Centralised MA Applications
Centralised MA procedures for applications submitted before the new Regulation's date of application, and still pending the day before that date, are to be completed under the old regime of Article 10 of Regulation (EC) No 726/2004.[14] The new Regulation will thus not apply to these procedures, even after its date of application.
3.4.2 Pending Post-Authorisation Studies
Procedures for imposed post-authorisation studies ("PASS") initiated under Article 10a of Regulation (EC) No 726/2004 before the new Regulation's date of application and pending the day before that date are completed in accordance with Article 20 of the new Regulation.[15] Unlike MA procedures, which complete under the old Regulation, pending PASS procedures switch to the new Regulation's Article 20 framework.
3.4.3 Renewals
For medicinal products authorised before the Regulation's application date, whose validity expires after that date, renewals follow the procedures in Article 17 of the new Regulation.[16] Products falling into this window, i.e., products authorised under the old framework but not yet renewed by the cut-over, will be renewed under the new procedural rules.
3.4.4 Regulatory Data Protection: The Filing-Date Rule
The new regulatory protection periods do not apply to reference medicinal products for which the MA application was submitted before the new Regulation's application date.[17] These rules only apply to medicinal products for which the MA application was submitted after the new Regulation's application date.
This is one of the most commercially consequential transitional rules. The new market protection architecture, including any changes to the baseline duration of regulatory data protection and the new extension logic of "(8)+(1)+(1)+(1)", applies only to products whose reference MA application is submitted on or after the full application date. Earlier filings are grandfathered into the Article 14(11) of Regulation 726/2004 old regime of "(8)+(2)+(1)".
3.4.5 Orphan Medicinal Products: Three Separate Rules
The orphan transitional provisions are more layered than those for standard products, and the three rules must be read together.
First, existing orphan designations granted before the new Regulation's application date and still in the community register and not yet granted an orphan MA corresponding to the designation, are deemed compliant and entered into the new register of designated orphan medicinal products. Designation continuity is therefore preserved automatically.[18]
Second, for those legacy orphan designations, the new seven-year validity period for orphan designations runs from the Regulation's application date. This means the clock on the new validity mechanism does not reach back to the original designation date, but restarts from the cut-over, which is favourable for sponsors with older designations.[19]
Third, the orphan market exclusivity periods of the new Regulation do not apply to orphan medicinal products where the orphan MA application was submitted before the new Regulation's application date.[20] Those products receive the 10-year exclusivity period under Article 8(1) of Regulation (EC) No 141/2000.
However, and this is crucial, this derogation is only applicable for the first (or initial) orphan MA granted for a certain active substance. Any other MA application for an orphan medicinal product with the same active substance submitted before the application date is subject to the new Regulation for market exclusivity, including under Articles 71(3) and 72.[21]
Article 71(3) of the new Regulation introduces the concept of a "global Orphan MA", meaning that there can only be one period of market exclusivity per active substance. Under the current regime, orphan MAHs can obtain multiple full periods of 10 years of orphan market exclusivity if the orphan MAH obtains an MA for one or more new therapeutic indications for different orphan conditions. Under the new regime, such a new MA for a new therapeutic indication for a different orphan condition (but for the same active substance) will not lead to a new 10-year period of market exclusivity but will follow the exclusivity of the first indication (if still applicable). The new MA for the new indication (for the same active substance) can however lead to an extension of 12 months of that initial period of market exclusivity under Article 72 of the new Regulation if the orphan MAH obtains the MA for the new therapeutic indications for a different orphan condition at least two years before the end of the exclusivity period.
The concept of "global orphan MA" and the new 12-month extension rule for new authorised indications instead of a new period of exclusivity of 10 years will therefore already apply to orphan MA applications submitted before the application of the new Regulation. At this stage, it is unclear whether the legislator intends this new system to apply to all orphan MAs applied for before the entry into force, or only the applications that are still pending on the cut-over moment (i.e. in Q4 2028). Article 180(7a) does not specify explicitly that it would only apply to applications that are still pending on the date of application; it only refers to applications submitted before the date of application of the new Regulation.
Our view is that the new Regulation will apply only to subsequent orphan MA applications that are pending on the date of application of the new Regulation because, if it were to apply to subsequent orphan MAs that have already been granted before the application date of the new Regulation – which have resulted in an additional 10-year period of exclusivity – this would undermine rights lawfully granted under the old regime, which could possibly be open to challenge before a court.
In that hypothesis, this means that, if a company holds multiple orphan designations for a certain active substance, they should consider accelerating the submission of the second (and subsequent) orphan MA dossier(s) before the entry into force of the new Regulation to be able to benefit from a second period of 10-year market protection:
- If the second orphan MA is acquired before the entry into force of the new Regulation (expected late 2026), the indication can benefit from a full period of 10-year market exclusivity.
- If the second orphan MA is applied for but not yet acquired before the date of application of the new Regulation, it will not benefit from an independent period of market exclusivity but it will follow the market exclusivity period of the first indication of that active substance in accordance with Article 71(3) of the new Regulation (if still under market exclusivity). In that hypothesis, it can however lead to a 12-month extension of the initial market exclusivity period.
3.4.6 Paediatric Investigation Plans ("PIP")
PIPs, waivers and deferrals granted under Regulation (EC) No 1901/2006 before the Regulation's application date are considered compliant, and procedures for PIP, waiver or deferral applications submitted before the application date are completed under the old regime of Regulation (EC) No 1901/2006.[22]
4. The New Directive
A directive is evidently of a different legal nature than a Regulation, meaning that it does not have direct effect (even after its date of application)[23], but it has to be transposed into national law. The "date of application" of the new Directive is therefore the date by which Member States must have the national law in place to apply the provisions of the new Directive.
Under Article 219(1) of the Directive, Member States must bring into force the laws, regulations and administrative provisions necessary to comply with the new Directive by 24 months after its date of entry into force and must apply those provisions from that same date. The date of entry into force is, as for the Regulation, 20 days after its publication in the OJEU. The new Directive has (unlike the new Regulation) a single date of application which is the date of publication + 20 days + 24 months.
There is one exception to the single application date, which is Article 56a regarding the launch and supply conditionality mechanism, which Member States may choose to apply earlier. That exception is addressed below, after we have explained the general transitional regime.
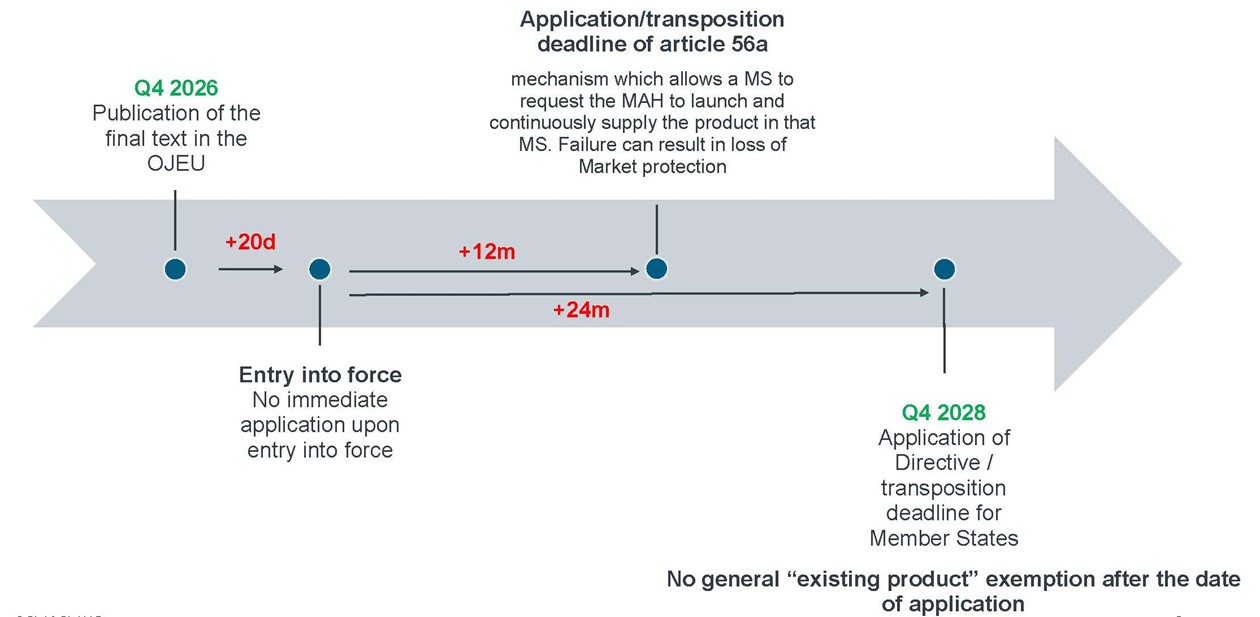
4.1 Impact of Application on Pending Applications and Procedures
No General "Existing Product" Exemption
The new Directive applies to medicinal products and to registrations of homeopathic and traditional herbal medicinal products authorised or registered under Directive 2001/83/EC before the Directive's application date.[24] There is no general "existing product" exemption, which means that the new Directive's ongoing obligations (pharmacovigilance, product information maintenance, shortage notification at national level, …) apply to the existing authorised portfolio from the application date.
Medicinal products placed on the market prior to the date of application, which do not comply with the requirements of the new Directive, may be marketed until the stocks of the medicinal products are exhausted.[25]
Exceptions - Pending MA and Pharmacovigilance Applications
National MA application procedures submitted under Article 19 of Directive 2001/83/EC before the new Directive's application date and pending the day before that date are to be completed under the regime of Directive 2001/83/EC.[26]
Procedures initiated on the basis of Articles 29, 30, 31 and 107i of Directive 2001/83/EC (covering mutual recognition, referrals, and pharmacovigilance union procedures), before the application date and that were pending on the day before that date are to be completed in accordance with the applicable provisions of Directive 2001/83/EC as they stood on the day before the application date.
This, too, can lead to asymmetrical periods of market protection if a particular company submitted a decentralised MAA in one set of countries before the date of application of the new Directive, and in another set of countries only after its date of application.
Regulatory Data Protection: The Filing-Date Rule
A reference medicinal product for which the MA application was submitted before the new Directive's application date remains subject to the data protection rules in Article 10 of Directive 2001/83/EC.[27]
The logic mirrors the Regulation's grandfathering for centrally authorised products. The trigger is the submission date of the reference product MA application relative to the application date of the Directive. Products whose reference application predates the cut-over retain the regime of Article 10 of Directive 2001/83/EC. Products whose reference application falls on or after the cut-over fall under the new regime of Article 81 of the new Directive. Portfolio segmentation around this date is therefore a priority exercise for any originator company with a mixed pipeline.
Renewals
For medicinal products authorised before the Directive's application date whose validity expires after that date, renewal follows the procedures in Article 46 of the new Directive.[28] The same forward-looking cut-over applies as under the Regulation. Renewal dossiers falling due after the application date must comply with the new Directive's Article 46 requirements, not those of Directive 2001/83/EC.
Labelling and package leaflet: A five-year continued marketing window
By derogation from Chapter VI of the new Directive, medicinal products placed on the market under Directive 2001/83/EC before the new Directive's application date may continue to be made available until five years after the application date, provided they comply with the labelling and package leaflet provisions of Title V of Directive 2001/83/EC as applicable on the day before the application date.[29]
Electronic Package Leaflet: Staggered Application Tied to Implementing Acts
In general, the requirement to make the package leaflet available electronically under Article 63(1) applies immediately for MA applications submitted after the Directive's application date.[30]
However, for medicinal products for which the MA application was submitted before the application date of the new Directive, the e-leaflet requirement applies three years after the Directive's application date, again conditional on adoption of the implementing act.
Medicinal products authorised before entry into force that do not comply with the e-leaflet requirement may continue to be placed on the market, distributed, dispensed, sold and used until stocks are exhausted. [31]
Reporting Obligations (Financial Support/R&D Costs): Non-Retroactivity for Existing Authorisations
Article 57 of the new Directive imposes an obligation on MAHs to declare to the public any direct financial support received from any public authority or publicly funded body, philanthropic organisation, or not-for-profit organisation or fund in relation to its R&D activities.
Medicinal products authorised under the old Directive before the cut-over are exempt from these financial transparency reporting obligations.[32] Medicinal products for which the MA application was filed before the date of application but that were only authorised after the date of application of the new Directive will need to comply.
Early Application of Article 56a: The Supply Obligation
Article 56a introduces the launch and supply conditionality mechanism, which allows a Member State, within one year of an MA grant, to request the MAH to launch and continuously supply the product in that Member State. The failure to comply within three years of such a request triggers a lapse of marketing protection in that Member State.
This mechanism may be applied by MS as from 12 months after the entry into force of the new Directive.[33] Note that this mechanism may also be invoked in relation to centrally authorised products (and thus before the date of application of the biggest part of the new Regulation).
A specific transitional interaction with the old framework is established for the application of this mechanism. Namely, if a medicinal product receives its MA during the gap period after entry into force of the new Directive but before full national transposition, it is exposed to the loss of the market protection under the old framework in any Member State that chooses to invoke Article 56a of the new Directive early, if the MAH does not comply.[34]
This means that a product granted an MA between the entry into force and the date of application of the new Directive becomes vulnerable to generic and biosimilar entry in that Member State if it does not comply with MS requests under Article 56a of the new Directive, notwithstanding that the new Directive does not apply yet for the remainder.
[2] Link to article: The EU Pharma Package Reaches a Milestone: Formal Adoption on the Horizon, Mette-Marie Henrichsen.
[3] Commission welcomes political agreement on major reform of EU pharmaceutical rules.
[4] 20 days after the publication in the OJEU.
[5] 6 or 12 months after the entry into force of the relevant provisions.
[6] See Article 71(3) new Regulation on the new concept of a "global orphan MA". Under Article 72 of the new Regulation, such new indication for a certain active substance could however give rise to a twelve-month prolongation of the market exclusivity, provided that it is covered by a separate orphan designation.
[7] See Article 181 of the new Regulation.
[8] There is also Article 141 on the competence of the EMA to communicate with other third country competent authorities and Article 154(5) on the EMA's operational expenditure.
[9] The MAH to whom the TEV is granted may transfer the RDP to another medicinal product of which it holds an EU MA or transfer it to another MAH in the EU.
[10] See Article 39 of Directive 2014/24/EU of the European Parliament and of the Council of 26 February 2014 on public procurement.
[11] See Part V of Annex IV of the new Regulation for detailed requirements of the shortage prevention plan.
[12] The "MSSG" is the Executive Steering Group on Shortages and Safety of Medicinal Products.
[13] Article 179 new Regulation.
[14] Article 180(2) new Regulation.
[15] Article 180(3) new Regulation.
[16] Article 180(3a) new Regulation.
[17] Article 180(4) new Regulation.
[18] Article 180(5) new Regulation.
[19] Article 180(7) new Regulation.
[20] Article 180(7a) new Regulation.
[21] Ibid.
[22] Article 180(9) new Regulation.
[23] Note that, by way of exception, certain clear provisions might be directly invoked by individuals in the absence of implementing legislation.
[24] Article 218(3) new Directive.
[25] Article 218(6b) new Directive.
[26] Article 218(1) new Directive.
[27] Article 218(5) new Directive.
[28] Article 218(6a) new Directive.
[29] Article 218(7) new Directive.
[30] However, this is only the case provided that the Commission implementing act pursuant to Article 63(6) of the new Directive is adopted.
[31] Article 218(8) new Directive.
[32] Article 218(6) new Directive.
[33] Article 219(1a) new Directive.
[34] Ibid.
/Passle/MediaLibrary/Images/2026-07-30-15-23-08-547-6a6b6c5cf10dbec0100718d8.jpeg)
/Passle/MediaLibrary/Images/2026-07-27-08-51-04-398-6a671bf8a3513a74a4f37b66.jpg)
/Passle/65b10a249576f7f0a5a2f163/MediaLibrary/Images/2026-06-11-14-33-24-680-6a2ac734e20430e1709e6e83.png)
/Passle/MediaLibrary/Images/2025-08-06-08-07-29-578-68930d41763c43c02079dea7.jpg)